

Pulmonary Arterial
Hypertension (PAH)
Preclinical Development of CM5480 for the Treatment of Pulmonary Arterial Hypertension
CM5480 has the potential to be a first-in-class, differentiated therapy for the treatment of PAH. Preclinical studies in PAH models have shown that CM5480 restored or improved multiple disease-affected pathways and functions—including heart contraction and cardiac output, gene expression profiles, DNA repair, and metabolism. Treatment with CM5480 also significantly reduced right ventricular dysfunction (RVD) both as a monotherapy and in combination with existing PAH therapies. These data support the further development of CM5480 as a potentially disease-modifying treatment in PAH.
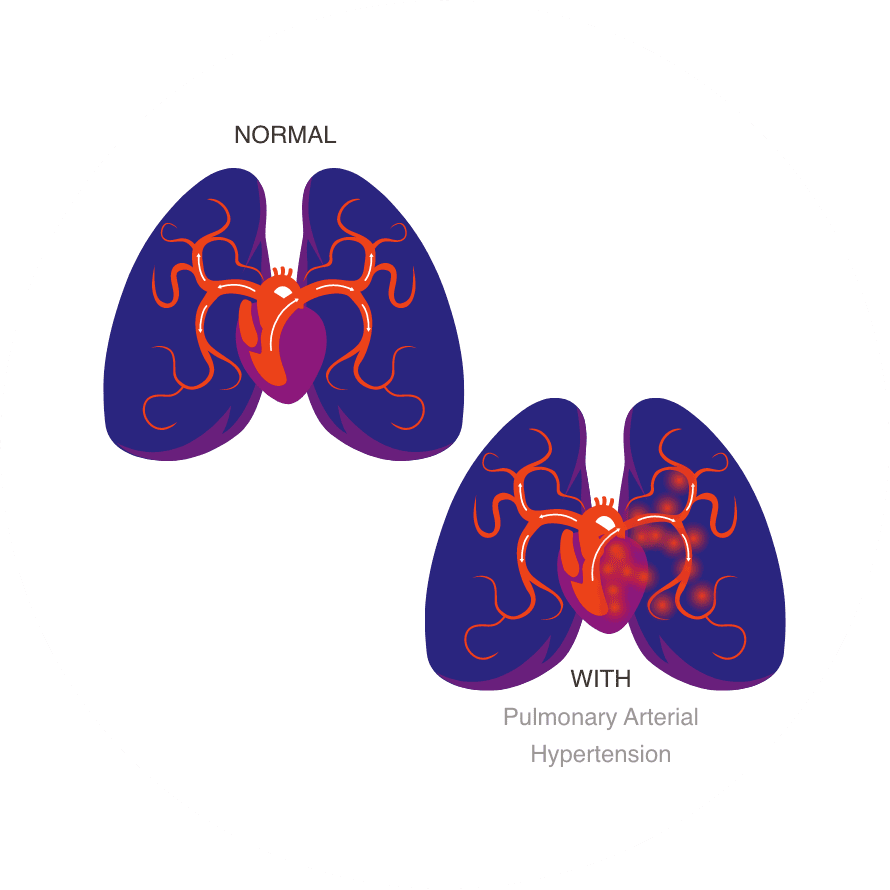
PAH is a rare pulmonary vascular disease characterized by progressive narrowing and thickening of pulmonary arteries, resulting in chronic elevation in pulmonary artery pressure and pulmonary vascular resistance (PVR). Over time, this can lead to right ventricular (RV) hypertrophy, RVD, RV failure (RVF), and death. Data have identified Orai1 CRAC channels as a shared, disease-driving mechanism linking PVR and RVF in PAH.
Current standard of care (SOC) therapies for patients with PAH target several key disease-related signaling pathways but do not address the Orai1 pathway. These SOC therapies are not curative and do not directly target RV function, which is the primary determinant of prognosis in PAH. It is important, therefore, to develop new treatment strategies targeting both RV function and pulmonary vascular disease. Previous preclinical studies have demonstrated that dysregulation of calcium entering pulmonary smooth muscle and endothelial cells through CRAC channels is a critical contributor to the pathogenesis of PAH and RVD. Furthermore, translational studies have found that Orai1 expression is increased in pulmonary arterial smooth muscle cells and pulmonary venous smooth muscle cells in patients with pulmonary veno-occlusive disease (PVOD), a rare form of PAH.
estimated number of adults with PAH in the US
- Chronic elevation in pulmonary artery pressure and pulmonary vascular resistance
- Right ventricular (RV) hypertrophy
- RV dysfunction
- RV failure
- Death
